Resources
Tools
The following tools are listed by OMICtools when searching by "exposome" (date: Feb 08, 2018)
- Public Databases
- Exposome Explorer: Exposome-Explorer contains detailed information on the nature of biomarkers, populations and subjects in which biomarkers have been measured, samples analysed, methods used for biomarker analyses, concentrations in biospecimens, correlations with external exposure measurements, and biological reproducibility over time.
- T3DB: The focus of the T3DB is on providing mechanisms of toxicity and target proteins for each toxin.
- R Packages
- rexposome: Permits to investigate the exposome and to proceed association analyses between exposures and health outcomes. rexposome accepts any clustering method that allows to obtain a classification of samples. The package offers a wide set of functions to preprocess exposome data. It provides several methods that allow (i) to transform the exposures, (ii) to check for normality in exposome, and (iii) to standardize the exposures, among others.
- MultiDataSet: MultiDataSet is designed to manage data from different omics experiment in the same individuals. The software contains methods to perform typical operations, such as sub-setting or selecting common samples, making integrative analysis more accessible to users with a low expertise in bioinformatics.
- Web Applications
- ChemRICH: ChemRICH is a chemical similarity enrichment analysis software for metabolomics datasets utilizing medical subject headings and Tanimoto substructure chemical similarity coefficients. It can cluster metabolites into non-overlapping chemical groups.
The tools listed here have been developed by: Clinical Metabolomics.
apLCMS, LC-MS metabolomics feature extraction
The apLCMS is used for preprocessing LC-MS data. The major improvements include the following aspects: the adaptive tolerance level searching, to apply non-parametric methods of fine-tune intensity grouping, to better preserve weak signals and the estimation method on peak intensities for absolute quantification. The algorithms are implemented in an R package apLCMS.
More about the tool: Yu, T., Park, Y., Johnson, J. M., and Jones, D. P. (2009). apLCMS—adaptive processing of high-resolution LC/MS data. Bioinformatics, 25(15), 1930-1936.
xMSanalyzer
xMSanalyzer is an automated pipeline for processing of metabolomics data which integrates with packages apLCMS and XCMS, but it could be also used to improve the data extraction steps performed by other software. It includes the following functions: data extraction, quality control assessment, detection of overlapping and unique metabolites in multiple datasets as well as batch annotation of metabolites.
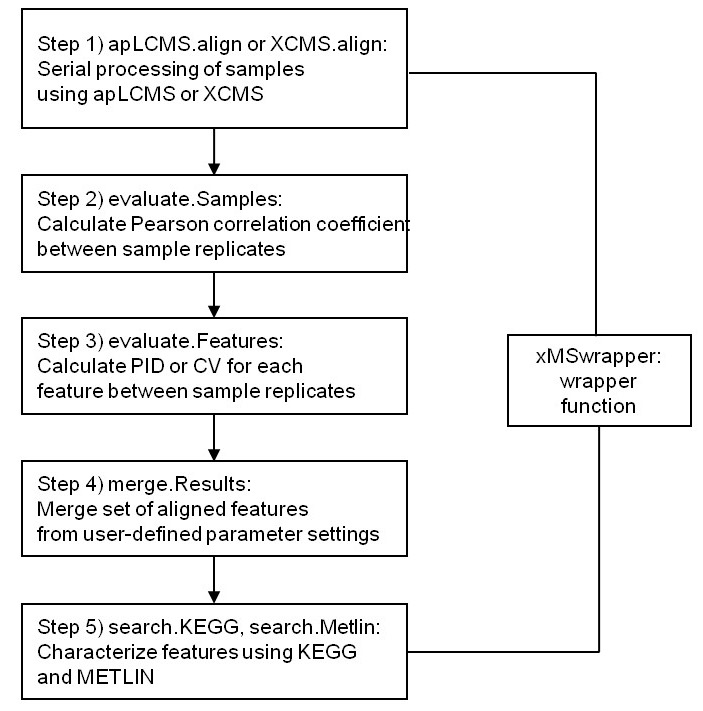
Fig 1. xMSanalyzer workflow (Taken from Uppal et al. 2013)
More about the tool: Uppal, K., Soltow, Q. A., Strobel, F. H., Pittard, W. S., Gernert, K. M., Yu, T., &emp Jones, D. P. (2013). xMSanalyzer: automated pipeline for improved feature detection and downstream analysis of large-scale, non-targeted metabolomics data. BMC bioinformatics, 14(1), 15.
Mummichog
Mummichog is a method to post-process high-throughput metabolomics data, namely, metabolite identification . This software features a biological activity prediction method which is directly from mass spectrometry data without priori identification of metabolites. Both gene expression and metabolite identification confirmed the predicted results.
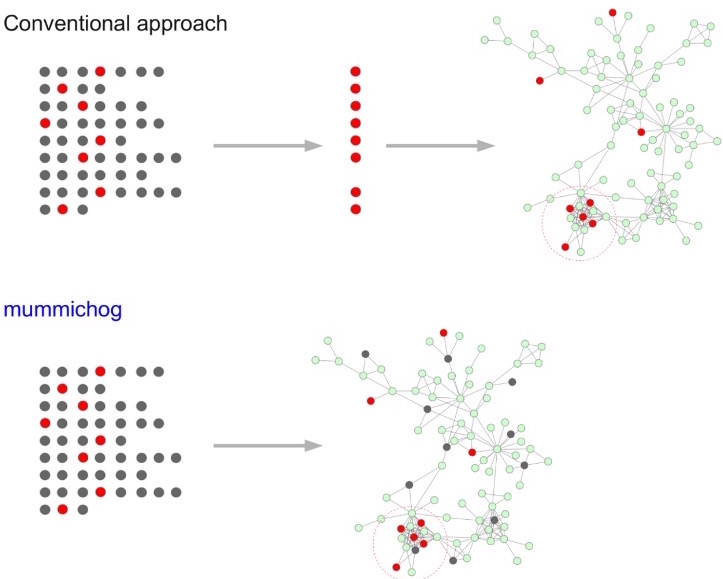
Fig 2. Workflow comparison between conventional methods VS Mummichog (Taken from Li et al. 2013)
More about the tool: Li, S., Park, Y., Duraisingham, S., Strobel, F. H., Khan, N., Soltow, Q. A., ... &emp Pulendran, B. (2013). Predicting network activity from high throughput metabolomics. PLoS computational biology, 9(7), e1003123.